
Dr. Eugene DePrince, Professor
Education and Training:
- 2004: BS in Chemistry, University of Tennessee, Knoxville
- 2009: PhD in Chemistry, University of Chicago
- 2010-11: Computational Post-Doctoral Fellow, Argonne National Laboratory
- 2011-13: NSF American Competitiveness in Chemistry Post-Doctoral Fellow, Georgia Institute of Technology
I received my undergraduate degree in chemistry from the University of Tennessee. In my sophomore year, I took a course in “chemical programming” that was my first introduction to programming in any kind of formal setting and kicked off my interest in the topic. (Fun fact: one of the only things I remember from this course is that, in the Perl programming language, the command print "\007" will make the computer beep.)
I joined the research group of the professor that taught the course (R.J. Hinde), where I developed computer codes to model interactions between light and metallic nanoparticles. For graduate school, I attended the University of Chicago, where I worked with Prof. David Mazziotti. My research focused on the development of quantum chemistry methods that relied on reduced density matrices as fundamental descriptors of electronic structure, as opposed to more familiar quantities like the density (which is used in density functional theory, DFT) or the wave function.
When did you join FSU? What made you choose your university to build your research program?
I joined the faculty of the Department of Chemistry & Biochemistry at FSU in 2013. From the time that I interviewed at FSU until now, I have been consistently impressed by FSU’s facilities and the caliber of my colleagues. Having lived in Chicago for six years for graduate school and a postdoc, I can also say that the Florida winters are amazing.
When did you become interested in quantum research? Who or what inspired you?
As an electronic structure theorist, pretty much all aspects of my research program are “quantum.” To be a bit more specific, I have long been interested in computational descriptions of light-matter interactions. In the last decade or so, a large number of experiments have shown that strong light-matter coupling in an optical cavity can result in a range of interesting and sometimes surprising phenomena, where the quantum nature of light can be important. On the theory side, there has been an explosion in interest in new multicomponent theories that model electron and photon degrees of freedom on equal footing within the framework of cavity quantum electrodynamics (QED). The opportunity to develop completely new theories does not come along every day, so, as someone with a passion for theory and software development, this has been an exciting time.
What are your current research interests? Could you give an example of some recent result that you feel especially passionate about?
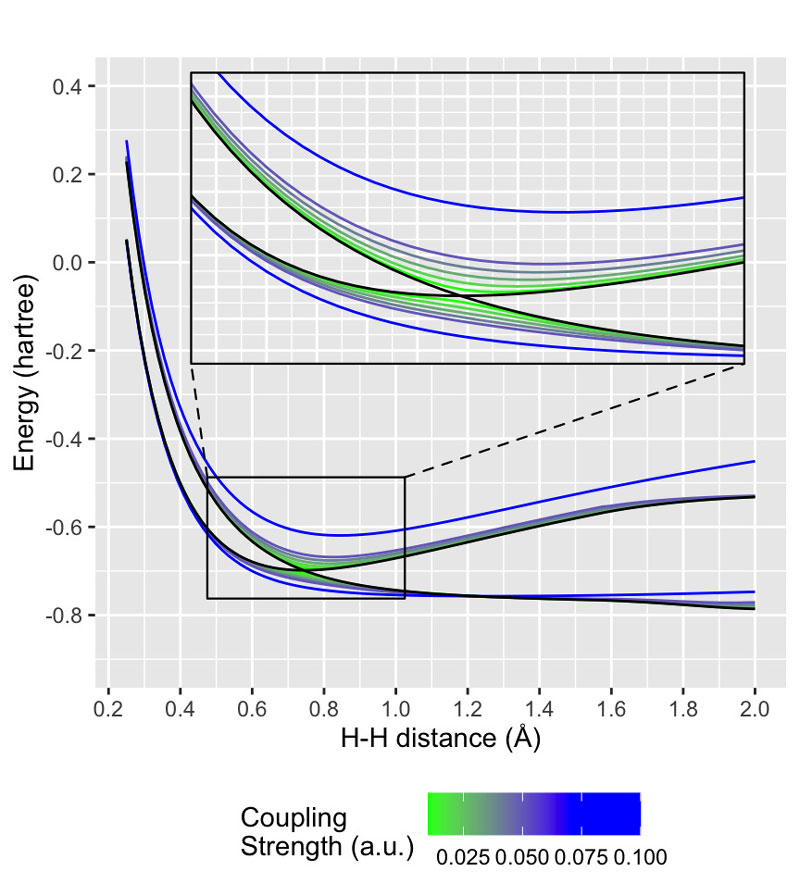
My group currently has two main research directions. On one hand, we develop cavity QED generalizations of familiar electronic structure methods to model strong interactions between electron and photon degrees of freedom. Using these approaches, we can make predictions regarding situations where embedding a molecule within an optical cavity can lead to interesting changes in the electronic structure of that molecule. We recently submitted a manuscript showing that second-order molecular response properties can be quite sensitive to optical cavity modes; for example, we showed the static first hyperpolarizability of a cavity-embedded molecule can change by up to 20%, as compared to that of the isolated molecule, at experimentally achievable coupling strengths.
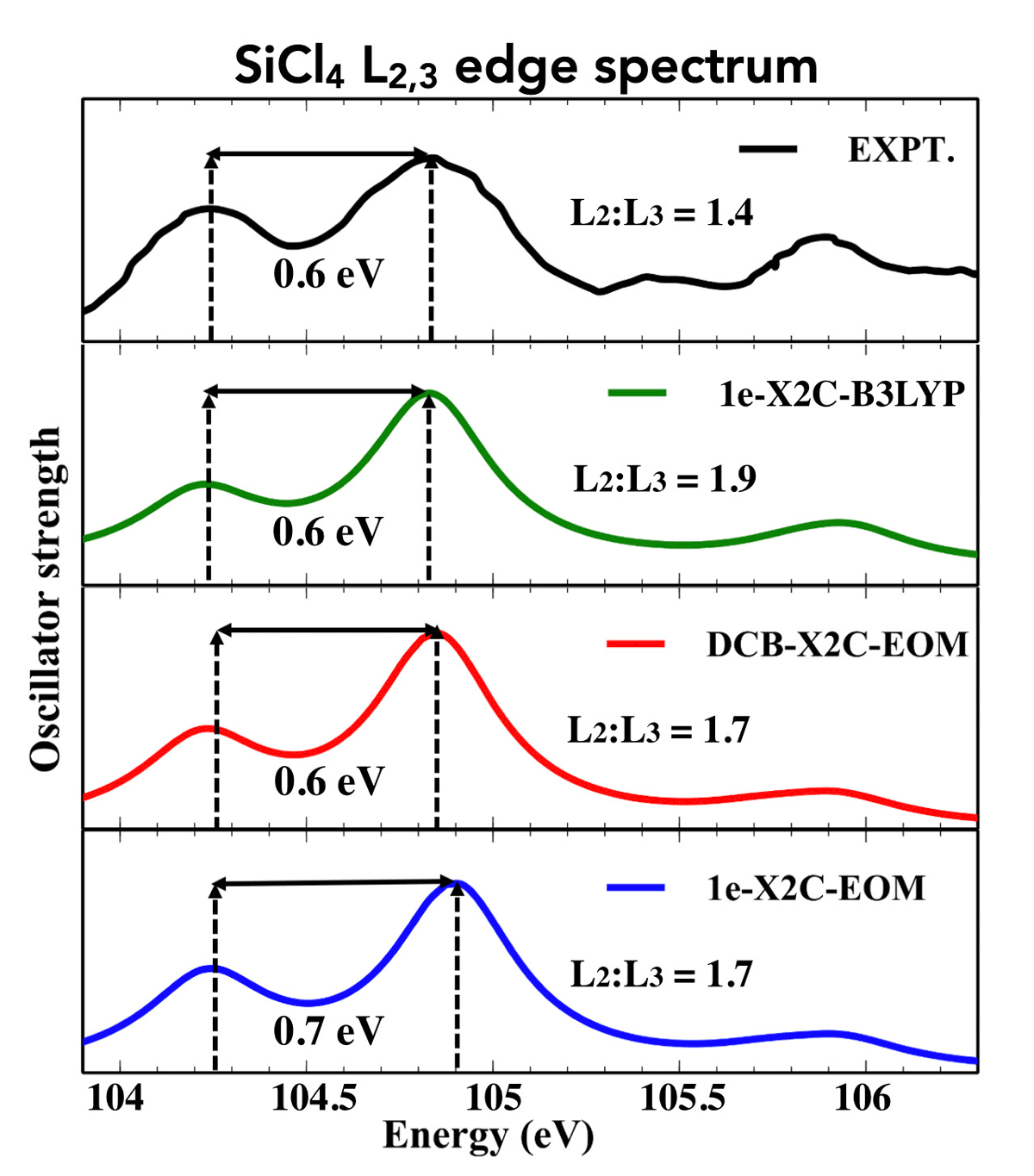
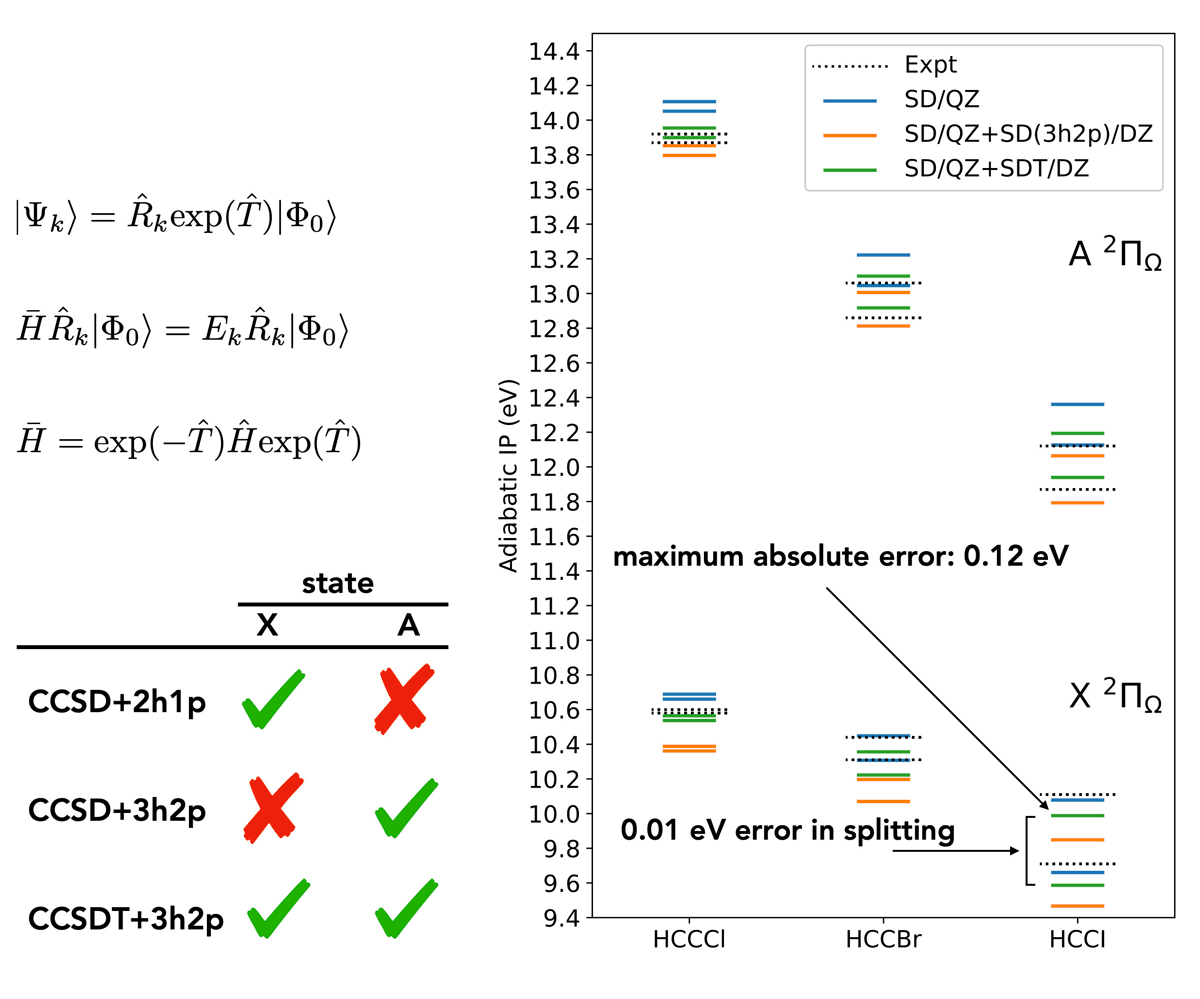
Another key research direction involves the development of high-accuracy electronic structure methods that capture spin-free and spin-dependent relativistic effects. The basic idea here is that we would like to be able to make quantitative predictions of electronic structure (e.g., molecular properties, energy levels, etc.) for systems where spin-orbit coupling effects cannot be ignored. As an example, we recently submitted a manuscript describing an approach evaluating double ionization potentials (IPs), which is the energy required to remove two electrons from an atom or molecule. The approaches we have developed are capable of predicting double IPs that are accurate within 0.1 eV, as compared to experimentally-obtained values. Splitting between different spin-orbit states of the doubly ionized species are even more accurate, often to the 0.001 eV level.
How would you describe your research to the general public? Why is your topic important?
We develop theories and computer software to model the behavior of electrons in molecules and materials. Simulating and understanding electronic structure is important because it allows us to make useful predictions of molecular energies, structures, or properties that can guide experimental studies in a laboratory. The simplest example would be that, given a chemical reaction, electronic structure calculations could make predictions about the relative energies of the reactants or products, as well any energetic barriers to the reaction. As a more specific “quantum” example, several research groups at FSU are interested in the design and manipulation of “molecular spin qubits,” which are molecules that could serve as the fundamental unit of information in quantum computing or other quantum applications. Electronic structure calculations can be used to predict the energies or lifetimes of the different electronic states that are relevant to the spin qubit, as well as how these properties change with different molecular structures. In this way, theoretical predictions can guide the synthesis of new spin qubit candidates with desirable properties.

What are your interests outside of research? What do you like to do in your free time?
Food is the best, and I really enjoy cooking. My children recently told me that I am not as good a cook as my father-in-law, though, because I do not catch fish. I fail to see the connection.